Identification of somatic and germline variants from tumor and normal sample pairs
Reproduced By Genomics Two A
Introduction
Mutations (random single or multiple base changes) in DNA or RNA can have a beneficial (eg in evolution), neutral or harmful effect in an organism. Many diseases including mostly cancers (second leading cuase of death) are as a result of harmful mutations in crucial genes eg Tumor suppressor genes, that cause cells to grow and divide uncontrollably, infiltrating and destroying normal body tissues. These mutations can be germline (inhrited) or somatic (acquired after birth), and a common kind of genetic mutation as a result of either is Loss of Heterozygosity (LOH). LOH usually leads to loss of one normal copy or a group of genes, which is a common even in cancer development. Germline mutations can easily be identified by comparing a sample genome to a reference, however the story is quite different when it comes to somatic mutations as we need both a normal and tumor tissue DNA from the patient.
In this project, we aimed at reproducing a workflow that identifies germline and somatic variants, variants affeted by LOH using both a health and tumor tissue, from which we would report variant sites and genes affected that could likely be the cause to the disease. Such insights can help us track the genetic events driving tumorigenesis in patients and might be useful in diagnosis, prognosis, developing and guiding therapeutics strategies.
Below is our Graphical abstract summarizing the key steps we took to achieve this.
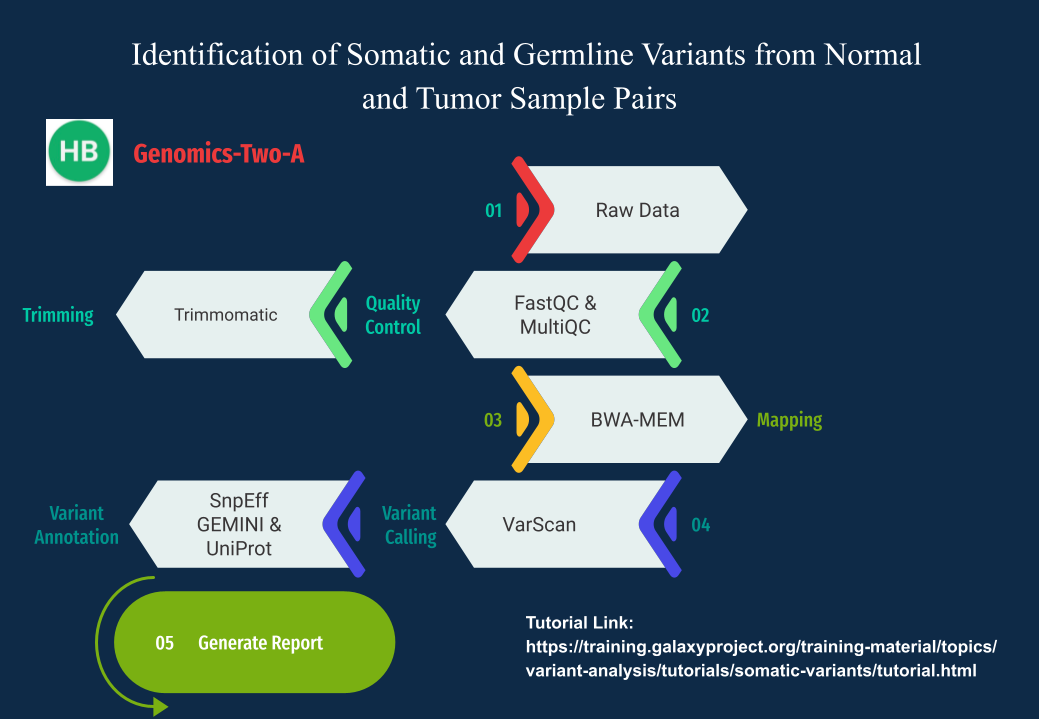 Figure 1: Graphical Abstract for Genomics-Two-A
Figure 1: Graphical Abstract for Genomics-Two-A
We reproduced this tutorial both as a Galaxy Tutorial as well as Linux Pipeline.
Go To Section:
- Introduction
- Section One: Linux Pipeline
- Section Two: Galaxy Workflow
- Contributors
Section One: Linux Pipeline.
## Dataset Description
The datasets used in this analysis (reads from human chromosomes 5, 12 and 17), were obtained from a cancer patient’s tumor and normal tissue samples. The normal tissue coudn't be the only sample used because healthy tissue contains many variants and every individual inherits a unique pattern of many variants from their parents. The samples (paired end) were two in number.
## Data download
The datasets were downloaded from Zenodo using the wget command.
### Target dataset
```
echo -e "\n Downloading data... \n"
mkdir -p raw_data
cd raw_data
wget https://zenodo.org/record/2582555/files/SLGFSK-N_231335_r1_chr5_12_17.fastq.gz
wget https://zenodo.org/record/2582555/files/SLGFSK-N_231335_r2_chr5_12_17.fastq.gz
wget https://zenodo.org/record/2582555/files/SLGFSK-T_231336_r1_chr5_12_17.fastq.gz
wget https://zenodo.org/record/2582555/files/SLGFSK-T_231336_r2_chr5_12_17.fastq.gz
```
### Reference sequence
```
echo -e "\n Downloading reference sequence... \n"
wget https://zenodo.org/record/2582555/files/hg19.chr5_12_17.fa.gz
#unzip reference
unzip hg19.chr5_12_17.fa.gz
```
## Pre-processing and Trimming
### i) Quality check.
The reads quality were examined using fastqc and an aggregate report generated by multiqc.
#### Description
"FastQC aims to provide a way to do quality control checks on sequence data. Within the `fastq` file is quality information that refers to the accuracy of each base call. This helps to determine any irregularies or features that make affect your results such as adapter contamination
#### Installation
```{bash}
conda install -c bioconda fastqc --yes
conda install -c bioconda multiqc --yes
```
#### Command
```bash
echo -e "\n Data Preprocessing... \n"
mkdir -p Fastqc_Reports #create directory for the fastqc output
```
```
#Qc on reads
for sample in `cat list.txt`
do
fastqc raw_data/${sample}*.fastq.gz -o Fastqc_Reports
done
multiqc Fastqc_Reports -o Fastqc_Reports
```
The multiqc report can be examined from [here](/Somatic-and-Germline-variant-Identification-from-Tumor-and-normal-Sample-Pairs/multiqc_report_linux.html).
From the report, the reads quality are great, a few adapters are however observed.
### ii) Removing low quality sequences using Trimmomatic
<http://www.usadellab.org/cms/?page=trimmomatic>
#### Description
`Trimmomatic` is a wrapper script that automate quality and adapter trimming. After analyzing data quality, the next step is to remove sequences that do not meet quality standards.
#### Installation
```
conda install -c bioconda trimmomatic --yes
```
#### Command
```
mkdir -p trimmed_reads
for sample in `cat list.txt`
do
trimmomatic PE -threads 8 raw_data/${sample}_r1_chr5_12_17.fastq.gz raw_data/${sample}_r2_chr5_12_17.fastq.gz \
trimmed_reads/${sample}_r1_paired.fq.gz trimmed_reads/${sample}_r1_unpaired.fq.gz \
trimmed_reads/${sample}_r2_paired.fq.gz trimmed_reads/${sample}_r2_unpaired.fq.gz \
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10:8:keepBothReads \
LEADING:3 TRAILING:10 MINLEN:25
fastqc trimmed_reads/${sample}_r1_paired.fq.gz trimmed_reads/${sample}_r2_paired.fq.gz \
-o trimmed_reads/Fastqc_results
done
multiqc trimmed_reads/Fastqc_results -o trimmed_reads/Fastqc_results
```
The parameters shown below were used during trimming:
* PE - paired end
* threads - number of cores assigned to the task,
* LEADING - remove lead bases with low quality of 3
* TRAILING - remove trailing bases with low quality of 10
* MINLEN - remove reads below 25 bases long
* ILLUMINACLIP - used to remove adapters
* _Trused3-PE - adapter_, _2 - Maximum mismatch count_, _30 - Accuracy of the match between the two ‘adapter ligated’ reads for PE palindrome read alignment_, _10 - Accuracy of the match between any adapter against a read_, _8 - Minimum length of adapter that needs to be detected (PE specific/ palindrome mode_
The post trimming multiqc report can be found [here](/Somatic-and-Germline-variant-Identification-from-Tumor-and-normal-Sample-Pairs/post_trim_multiqc_report_linux.html). It is evident from the report that the quality of the reads improved having per base quality scores above 35 and no adapters observed. After trimming an average of 0.73% normal reads and 1.24% tumor reads were lost.
**NB: To view the multiqc html reports download the files and view them from your browser.**
## Mapped read postprocessing
### Description
Mapping of sample sequences against the reference genome is conducted with an aim of determining the most likey source of the observed sequencing reads.
`BWA-MEM` was used for alignment. The results of mapping is a sequence alignment map (SAM) format. The file has a single unified format for storing read alignments to a reference genome.
### Installation
```
conda install -y -c bioconda bwa
conda install -c bioconda samtools
conda install -c bioconda bamtools
```
### Command
#### Alignment
```
mkdir Mapping
#Index reference file
bwa index hg19.chr5_12_17.fa
#Perform alignment
bwa mem -R '@RG\tID:231335\tSM:Normal' hg19.chr5_12_17.fa trimmed_reads/SLGFSK-N_231335_r1_paired.fq.gz \
trimmed_reads/SLGFSK-N_231335_r2_paired.fq.gz > Mapping/SLGFSK-N_231335.sam
bwa mem -R '@RG\tID:231336\tSM:Tumor' hg19.chr5_12_17.fa trimmed_reads/SLGFSK-T_231336_r1_paired.fq.gz \
trimmed_reads/SLGFSK-T_231336_r2_paired.fq.gz > Mapping/SLGFSK-T_231336.sam
```
#### Conversion of the SAM file to BAM file, sorting and indexing
A Binary Alignment Map (BAM) format is an equivalent to sam but its developed for fast processing and indexing. It stores every read base, base quality and uses a single conventional technique for all types of data.
The produced BAM files were sorted by read name and indexing was done for faster or rapid retrieval. At the end of the every BAM file, a special end of file (EOF) marker is usually written, the samtools index command also checks for this and produces an error message if its not found.
```
for sample in `cat list.txt`
do
Convert SAM to BAM and sort it
samtools view -@ 20 -S -b Mapping/${sample}.sam | samtools sort -@ 32 > Mapping/${sample}.sorted.bam
Index BAM file
samtools index Mapping/${sample}.sorted.bam
done
```
#### Mapped reads filtering
```
for sample in `cat list.txt`
do
#Filter BAM files
samtools view -q 1 -f 0x2 -F 0x8 -b Mapping/${sample}.sorted.bam > Mapping/${sample}.filtered1.bam
done
```
To view the output of the results use :
```
samtools flagstat
```
#### Duplicates removal
During library construction sometimes there's introduction of PCR (Polymerase Chain Reaction) duplicates, these duplicates usually can result in false SNPs (Single Nucleotide Polymorphisms), whereby the can manifest themselves as high read depth support. A low number of duplicates (<5%) in good libraries is considered standard.
```
#use the command
for sample in `cat list.txt`
do
samtools collate -o Mapping/${sample}.namecollate.bam Mapping/${sample}.filtered1.bam
samtools fixmate -m Mapping/${sample}.namecollate.bam Mapping/${sample}.fixmate.bam
samtools sort -@ 32 -o Mapping/${sample}.positionsort.bam Mapping/${sample}.fixmate.bam
samtools markdup -@32 -r Mapping/${sample}.positionsort.bam Mapping/${sample}.clean.bam
done
#or
samtools rmdup SLGFSK35.sorted.bam SLGFSK35.rdup and samtools rmdup SLGFSK36.sorted.bam SLGFSK36.rdup.
```
#### Left Align BAM
```
for sample in `cat list.txt`
do
#-c -> compressed, -m -> max-iterations
cat Mapping/${sample}.clean.bam | bamleftalign -f hg19.chr5_12_17.fa -m 5 -c > Mapping/${sample}.leftAlign.bam
```
#### Recalibrate read mapping qualities
```
for sample in `cat list.txt`
do
samtools calmd -@ 32 -b Mapping/${sample}.leftAlign.bam hg19.chr5_12_17.fa > Mapping/${sample}.recalibrate.bam
done
```
#### Refilter read mapping qualities
```
for sample in `cat list.txt`
do
bamtools filter -in Mapping/${sample}.recalibrate.bam -mapQuality "<=254" > Mapping/${sample}.refilter.bam
done
```
## Variant calling and classification
<http://varscan.sourceforge.net/somatic-calling.html>
### Description
To be able to identify variants from the mapped samples, the tool `VarScan somatic` was used.
The command expects both a normal and tumor sample in `Samtools pileup` format and outputs an indel file and snp file.
The command reports germline, somatic, and LOH events at positions where both normal and tumor samples have sufficient coverage
### Installation
```
wget https://sourceforge.net/projects/varscan/files/VarScan.v2.3.9.jar
```
### Command
#### Convert data to pileup
```
mkdir Variants
for sample in `cat list.txt`
do
samtools mpileup -f hg19.chr5_12_17.fa Mapping/${sample}.refilter.bam --min-MQ 1 --min-BQ 28 \
> Variants/${sample}.pileup
done
```
#### Call variants
```
java -jar VarScan.v2.3.9.jar somatic Variants/SLGFSK-N_231335.pileup \
Variants/SLGFSK-T_231336.pileup Variants/SLGFSK \
--normal-purity 1 --tumor-purity 0.5 --output-vcf 1
```
#### Merge vcf
VarScan generates 2 outputs (indel.vcf and snp.vcf), merge the two into one vcf file using `bcftools.`
```
#merge vcf
bgzip Variants/SLGFSK.snp.vcf > Variants/SLGFSK.snp.vcf.gz
bgzip Variants/SLGFSK.indel.vcf > Variants/SLGFSK.indel.vcf.gz
tabix Variants/SLGFSK.snp.vcf.gz
tabix Variants/SLGFSK.indel.vcf.gz
bcftools merge Variants/SLGFSK.snp.vcf.gz Variants/SLGFSK.indel.vcf.gz > Variants/SLGFSK.vcf
```
## Variant Annotation
### Functional Annotation using `SnpEff`
<https://pcingola.github.io/SnpEff/examples/>
#### Description
`SnpEff` is a variant annotator and functional effect predictor. The output is appended to the vcf file with the field `ANN`. A snpEff database is required prior to performing annotation. In case the organism of interest is not present in the snpEff database, you can build the database using the snpEff command. If the organism is present in the database, download it using the snpEff command.
#### Installation
```
#download jar file
wget https://snpeff.blob.core.windows.net/versions/snpEff_latest_core.zip
# Unzip file
unzip snpEff_latest_core.zip
#download snpEff database
java -jar snpEff.jar download hg19
```
#### Command
```
#annotate variants
java -Xmx8g -jar snpEff/snpEff.jar hg19 Variants/SLGFSK.vcf > Variants/SLGFSK.ann.vcf
```
### Clinical Annotation using `gemini`
<https://gemini.readthedocs.io/en/latest/content/preprocessing.html>
#### Description
#### Installation
```
wget https://raw.github.com/arq5x/gemini/master/gemini/scripts/gemini_install.py
python gemini_install.py /usr/local /usr/local/share/gemini
```
#### Command
```
gemini load -v Variants/SLGFSK.ann.vcf -t snpEff Annotation/gemini.db
```
# Section Two: `GALAXY WORKFLOW` .
## 1. Data Preparation:
The sequencing reads that were used for analysis were obtained from a cancer patient's normal and tumor tissues.
There were a total of four samples. A forward reads sample and a reverse reads sample was obtained for both the normal and tumor tissue. A human reference genome, hg19 version was also used for analysis.
The first step was to create a new workflow we named Genomics_2_A in the galaxy window. We then imported the 4 fastq files and the reference using the `Upload` button then `Paste/Fetch Data` and pasting the corresponding links to the data, selecting datatype as `fastqsanger.gz` for the fsatq samples and `fasta` for the reference. Once done uploading, the attributes of the samples were edited using the pencil mark to **tumor** and **normal** correspondily for easier identification.
## 2. Quality Control & Check:
• FastQC: is a quality control tool for high throughput sequence data that gives a summary report about the sequence.
• MultiQC: A modular tool to aggregate results from bioinformatics analyses across many samples into a single report.
The MultiQC Output & Report:
For the quality control analysis, the following figures show that the chosen dataset is of high quality for both Normal R1 & R2 datasets and both Tumor R1 & R2 datasets even the tumor ones are of poorer quality than the normal ones:
o All nucleotides have high-quality scores, (the forward and reverse reads of normal and tumor patient’s tissues), as they all are present in high/good quality region.

o Good quality score distribution as the mean is high with a sharp, distinct peak.

o The actual mean of the GC% content is lower than the theoretical one and that is a non-normal distribution which may indicate some contamination; however, this peculiar bimodal distribution is considered to be a hallmark of the captured method as using Agilent’s SureSelect V5 technology for exome enrichment.

o The N content is zero, thus not indicating any bad base detection.

o Sequence duplication level is good as well with almost no PCR biases when library was prepared as there is no over-amplified fragment.

o There is a little presence of adaptors in the sequence.

## Read Trimming and Filtering
The next process after quality control is Trimming and Filtering. This process further helps to trim and filter raw reads to give a more improved quality.
° Tool - TRIMMOMATIC (Galaxy Version 0. 38. 0) is a fast, multithreaded command line tool that can be used to trim and crop low quality reads and remove the present adaptors in the reads to improve the quality of these processing datasets.
° Input data - The forward read FASTQ file (r1) and the reverse read FASTQ file (r2) of the normal tissue were run concurrently as paired-end by performing initial ILLUMINACLIP step.
° Parameters -


° Output data - Trimmed forward and reverse reads for each normal and tumor tissue.
- Orphaned forward and reverse reads for each normal and tumor tissue which the corresponding mate got dropped because of insufficient length after trimming. These datasets are empty and therefore deleted.
° The MultiQC Output & Report after Trimming:
The quality reads did not change much as the datasets were already of high quality although a small fraction of adapter is successfully removed as shown in the following report:






## Read Mapping
Once the sequence reads have been filtered and trimmed, read mapping or alignment is the next step in the bioinformatic pipeline. Read mapping is the process of aligning a set of reads to a reference genome to determine their specific genomic location. Several tools are available for read mapping but BWA-MEM (Galaxy Version 0.7.17.2) was used for our analysis as it is faster, accurate and supports paired-end reads. Read mapping was performed independently for the normal and tumor tissue using thesame parameters except otherwise stated.
Paramaters
- Locally cached human hg19 reference genome, Paired end reads, Forward and reverse trimmed reads (output of trimmomatic), Set read groups (SAM/BAM specification), auto-assign (no), Read Group Identifier (231335 for normal tissue and 231336 for tumor tissue), Read group sample name (normal for normal tissue and tumor for tumor tissue),
- For parameters not listed, default setting was used.
## Mapped reads postprocessing
### Mapped reads filtering
The tool used was a BAM tools filter called:  available on Galaxy. It produces newly filtered BAM datasets and only retains reads mapped to the reference successfully and have a minimal mapping quality of 1 and for which the mate read has also been mapped.
The quality of the output data is controlled by a series ofconditions and filters.
The BAM tools filter was run with these parameters:
The BAM datasets we filtered were:
1. The output of Map with BWA (mapped reads in BAM format)
2. The output of Map with BWA (mapped reads in BAM format)
The quality of the output data is controlled by a series of conditions and filters.
The Conditions set were as below:
The first filter involved selecting a BAM property to filter on which was mapQuality+ the filter on read mapping quality (on a phred scale):>=1
The mapping quality scale quantifies the probability that a read was misplaced.
The second filter involved selecting another BAM property to filter,for which we selected:isMapped(for mapped reads)+Selected mapped reads>Yes
The third filter involved selecting yet another BAM property to filter for which we selected isMateMapped (for paired-end reads with long inserts)+a confirmation to select mapped reads>yes
The last condition set involving opting to set rules for which we selected >No 
Then we ran the job. This was done for both the normal and tumor tissue data thus resulting in two datasets in the output results
## Duplicate Reads Removal
Tool: 
#### Significance
RmDup is a tool that identifies PCR duplicates by identifying pairs of reads where multiple reads align to the same exact start position in the genome. PCR duplicates arise from multiple PCR products from the same template binding on the flow cell.These are usually removed because they can lead to false positives
The read pair with the highest mapping quality score is kept and the other pairs are discarded.
It is important to note that this tool does not work for unpaired reads(in paired end mode) or reads that would pair where each maps to different chromosomes.
We used filtered reads datasets(BAM file) from the normal and the tumor tissue data- *outputs of Filter BAM datasets on a variety of attributes* .
We run RmDup on the following parameters:and The result was two new datasets in BAM format.The duplicate rate for both sets was well below 10% which is considered good.The tool standard error reflected the results below of unmatched pairs on chr5 and chr12 that otherwise were not included in the output data.

### Left-align reads around indels
The first Step in this is running the BamLeftAlign tool from the Tools set available on Galaxy. Then we have chosen the source for the reference genome as Locally cached and selected the filtered and dedicated reads datasets from the normal and the tumor tissue data which were the outputs of RmDup. Then we used Human: hg19 aa the genome reference and set the maximum number of iterations as 5, keeping all other settings as default and finally this will generate two new datasets, that is,one for each of the normal and tumor data.
### Recalibrate read mapping qualities
The next step after Left aligning the reads around indels is Recalibrating the read mapping qualities.
•RECALIBRATE READ QUALITY SCORES :
The first Step in Recalibrating read mapping qualities is running CalMD tool from Galaxy tool set. Firstly we have selected the left-aligned datasets from the normal and the tumor tissue data; the outputs of BamLeftAlign tool as the input for the BAM file to recalculate. Them we chose the source of reference genome as Use a built in genome as the required hg 19 reference genome was already in built in the Galaxy version we were using. We chose Advanced options as the choice for Additional options and we selected 50 as the Coefficient to cap the mapping quality of poorly mapped reads. And finally this step would produce two new datasets, that is one for each of the normal and tumor data.
### Refilter reads based on mapping quality
Eliminating reads with undefined mapping quality
We ran Filter BAM datasets on a variety of attributes tool using some parameters. The recalibrated datasets from the normal and the tumor tissue data which were the outputs of CalMD were selected as the BAM datasets to filter. Then we applied certain conditions as the options , in Filter, we selecte the MapQuality as the BAM property to Filter. Then set the value of less than or equal to 254 (<=254) as the Filter on read mapping quality (phred scale).
## Adding genetic and clinical evidence_based annotation : Creating a GEMINI database for variants
The next step after adding functional annotation to the called variants was to add "genetic and clinical based annotations"
The processes of the step will help observe more information on the variants like: the clinical and genetic aspects, prevalence in the population and the frequency of occurency.
Firstly, the output from function annotation was loaded into the GEMINI database so as to create a Gemini database where further annotations can be effectively carried
out.
The following optional contents of the variant were loaded as well: GERP scores- these are scores gotten from the constrained elements in multiple alignments by
quantifying the substitution deficits, CAAD scores, [N:B-high CAAD and GERP scores were observed in all pathogenicity components], gene tables, sample genotypes and
variant INFO fields.
These loaded variants in the gemini database are then annoatated by GEMINI annotate; this tool adds more explicit information on the output of VarScan that could not be indentified by Gemini load.
## Making variant call statistics accessible
Hence, we used Gemini annotate to extract three values : Somatic Status(SS), Germline p-value (GPV) and Somatic p-value(SPV) from the info generated by VarScan and added them to the Gemini database.
In order to crosscheck if all information extracted by the GEMINI database are in relation to variants observed in the population, we decided to annote by adding more information from the Single Nucleotide Polymorphism Database(dbSNP), also from Cancer Hotspots, links to CIViC database as well as more information from the Cancer Genome Interpreter (CGI)
For dbSNP: the last output from Gemini annotate was annotated with the imported dbSNP using the Gemini annotate tool. This process extrated dbSNP SNP Allele Origin (SAO) and adds it as "rs_ss" column to the existing database.
For Cancerhotspots: the last ouput generated from annotating dbSNP information was then annotated using GEMINI annotate tool, using the imported cancer hotspots as annotation source to extract "q-values" of overlapping cancerhotspots and add them as "hs_qvalue" column to the existing database.
Links to CIVic: the output of the last Gemini annoate for cancerhotspots was annotated using the imported CIViC bed as annotation source. We extracted 4 elements from this source and again added them as a list of "overlapping_civic_urls" to the existing Gemini database.
For the Cancer Genome Interpreter: the last output with the extracted infomation linking to CIViC was further annotated using the tool Gemini annotate with the imported CGI variants as an annotation source. the information extracted was recorded in the Gemini database as "in_cgidb" being used as the column name.
## Reporting Selected Subsets of Variants with GEMINI Query
GEMINI query syntax is built on the SQLite dialect of SQL. This query language enables users express different ideas when exploring variant datasets, for this analysis four (4) stepwise GEMINI queries were carried out.
1. A query to obtain the report of bona fide somatic variants
“GEMINI database”: the fully annotated database created in the last GEMINI annotate step i.e., Cancer Genome Interpreter(CGI)
“Build GEMINI query using”: *Basic variant query constructor*
“Insert Genotype filter expression”: ```gt_alt_freqs.NORMAL <= 0.05 AND gt_alt_freqs.TUMOR >= 0.10```
This genotype filter aims to read only variants that are supported by less than 5% of the normal sample, but more than 10% of the tumor sample reads collectively
“Additional constraints expressed in SQL syntax”: ```somatic_status = 2```
This somatic status called by VarScan somatic is one of the information stored in the GEMINI database.
By default, the report of this run would be output in tabular format; and a column header is added to it output.
The following columns were selected
* “chrom”
* “start”
* “ref”
* “alt”
* “Additional columns (comma-separated)”: ```gene, aa_change, rs_ids, hs_qvalue, cosmic_ids```
These columns are gotten from the variants table of the GEMINI database.
2. This second step has the same settings as the above step except for:
* “Additional constraints expressed in SQL syntax”: ```somatic_status = 2 AND somatic_p <= 0.05 AND filter IS NULL```
3. Run GEMINI query with same settings as step two, excepting:
* In “Output format options”
“Additional columns (comma-separated)”: ```type, gt_alt_freqs.TUMOR, gt_alt_freqs.NORMAL, ifnull(nullif(round(max_aaf_all,2),-1.0),0) AS MAF, gene, impact_so, aa_change, ifnull(round(cadd_scaled,2),'.') AS cadd_scaled, round(gerp_bp_score,2) AS gerp_bp, ifnull(round(gerp_element_pval,2),'.') AS gerp_element_pval, ifnull(round(hs_qvalue,2), '.') AS hs_qvalue, in_omim, ifnull(clinvar_sig,'.') AS clinvar_sig, ifnull(clinvar_disease_name,'.') AS clinvar_disease_name, ifnull(rs_ids,'.') AS dbsnp_ids, rs_ss, ifnull(cosmic_ids,'.') AS cosmic_ids, ifnull(overlapping_civic_url,'.') AS overlapping_civic_url, in_cgidb```
## Generating Reports of Genes Affected by Variants
In this step, gene-centred report is generated based on the same somatic variants we selected above.
As in the previous step we run GEMINI query but in advanced mode
• “Build GEMINI query using”: *Advanced query constructor*
• “The query to be issued to the database”: ```SELECT v.gene, v.chrom, g.synonym, g.hgnc_id, g.entrez_id, g.rvis_pct, v.clinvar_gene_phenotype FROM variants v, gene_detailed g WHERE v.chrom = g.chrom AND v.gene = g.gene AND v.somatic_status = 2 AND v.somatic_p <= 0.05 AND v.filter IS NULL GROUP BY g.gene```
However the “Genotype filter expression”: ```gt_alt_freqs.NORMAL <= 0.05 AND gt_alt_freqs.TUMOR >= 0.10``` remains the same
## Adding additional Annotation to the Gene-Centered Report
The aim of including extra annotations to the GEMINI-generated gene report (that is, the output of the last GEMINI query) is to make interpreting the final output easier. While GEMINI-annotate allowed us to add specific columns to the table of the database we created, it does not allow us to include additional annotations into the tabular gene report.
By simply using the Join two files tools on Galaxy, this task was achieved. After which, irrelevant columns were removed by specifying the columns that are needed. Three step wise process were involved here: One, we pulled the annotations found in Uniprot cancer genes dataset; second, we used the output of the last Join operation, annotated the newly formed gene-centered report with the CGI biomarkers datasets; and three, we used the output of the second Join operation, add the Gene Summaries dataset. Lastly, we ran Column arrange by header name to rearrange the fully-annotated gene-centered report and eliminate unspecified columns.
The last output of the Join operation was selected in the “file to arrange” section. The columns to be specified by name are: gene, chrom, synonym, hgnc_id, entrez_id, rvis_pct, is_TS, in_cgi_biomarkers, clinvar_gene_phenotype, gene_civic_url, and description. The result gotten was a [tabular gene report](https://github.com/Fredrick-Kakembo/Somatic-and-Germline-variant-Identification-from-Tumor-and-normal-Sample-Pairs/blob/main/Galaxy54-%5BColumn_arrange_on_data_53%5D%20(1).tabular), which was easy to understand and interpret.
---
## List of team members according to the environment used:
1. Galaxy Workflow:
- @Rachael - Adding genetic and clinical evidence-based annotations [Link to galaxy workflow](https://usegalaxy.eu/u/rachael-eo/w/workflow-constructed-from-history-genomics-twoarachael-1)
- @Mercy
- @Orinda
- @Heshica - Mapped Read Postprocessing (Left-align reads around indels , Recalibrate read mapping qualities and Refilter reads based on mapping quality)[Link to Galaxy Workflow](https://usegalaxy.eu/u/heshica_battina_chowdary/w/normal-and-tumor-analysisheshica-genomics-2a)
- @VioletNwoke - Read mapping [Link to galaxy workflow](https://usegalaxy.eu/u/violet/w/workflow-constructed-from-history-hackbiogenomicstwoaviolet-4)
- @AmaraA
- @Amarachukwu -Reporting Selected Subsets of Variants and Generating Reports of Genes Affected by Variants(GEMINI Query) [Link to Galaxy workflow](https://usegalaxy.eu/u/amara_chike/w/somatic-variant-tutorial-genomics-2-a-1)
- @Mallika
- @Olamide - Read Trimming and Filtering [Link to Galaxy Workflow](https://usegalaxy.eu/u/olamide21/w/identification-of-somatic-and-germline-variants-from-tumor-and-normal-sample-pairs)
- @NadaaHussienn - Quality Control and Check [Link to Galaxy Workflow](https://usegalaxy.eu/u/nadahussien/w/workflow-constructed-from-history-identification-of-somatic-and-germline-variants-from-tumor-and-normal-sample-pairs-3)
- @Christabel- [link to galaxy workflow](https://usegalaxy.eu/u/christabelmn1/w/somatic-and-germline-variants-and-gene-mutation-2)
- @Marvellous - [Workflow 1](https://usegalaxy.eu/u/marvellous_oyebanjo/w/workflow-constructed-from-history-identification-of-somatic-and-germline-variants-from-tumor-and-normal-sample-pairs) and [Workflow 2](https://usegalaxy.eu/u/marvellous_oyebanjo/w/workflow-constructed-from-history-identification-of-somatic-and-germline-variants-from-tumor-and-normal-sample-pairs-2)
- @juwon - Introduction
2. Linux Workflow
- @Praise
- @Fredrick
- @RuthMoraa
- @Kauthar - Preprocessing(pre/post trim qc) and read trimming.
- @Gladys
- @Nanje